


FASTQファイルの品質管理と前処理は、下流のデータ解析において高品質で信頼性の高いデータを取得するために重要なステップです。
本記事では、アダプター配列の除去やクオリティの低いリードの除去など、FASTQファイルを高速に前処理するツールであるfastpの使い方を解説します。
macOS Ventura 13.4
docker desktop : Version 4.20.1
fastp : Version 0.23.4
fastpとは
fastpは、FASTQファイルを高速に前処理するために作られたツールです。高いパフォーマンスを実現するためにマルチスレッドがサポートされています。
クオリティチェックにはFastQC、アダプター除去などのフィルタリングにはCutadaptなどのツールが存在しますが、fastpを使用することで、品質管理からフィルタリングまでを1つのツールで高速に完了させることができます。
fastpの使い方
fastpのdockerイメージ
dockerイメージは下記のリンクのものを使用します。今回使用するv0.23.4のバージョンのdockerイメージに良いものが見当たらなかったため、自分でdockerイメージを作成しました。
FASTQファイルの準備
fastpの動作確認に必要なFASTQファイルをダウンロードします。既に手元にある方は飛ばしてもらって大丈夫です。
シングルエンドでシーケンスされたデータとペアエンドでシーケンスされたデータの2種類を用意します。
シングルエンド
SRR24464805をダウンロードします。
docker container run --rm -v $PWD:/output -w /output pegi3s/sratoolkit:3.0.5 fasterq-dump -e 4 -p SRR24464805ペアエンド
SRR24920875をダウンロードします。
docker container run --rm -v $PWD:/output -w /output pegi3s/sratoolkit:3.0.5 fasterq-dump -e 4 -p SRR24920875これで、FASTQファイルの準備ができたので、早速fastpを使って前処理をしていきます。
fastpの基本形
基本的な使い方は、以下のように入力ファイルと出力ファイルを指定します。gzip圧縮の入出力にも対応しています。
シングルエンドの場合は、-iオプションで入力ファイル、-oオプションで出力ファイルを指定します。FASTQファイルの拡張子は「.fq」とも書けます。
fastp -i SRR24464805.fastq -o SRR24464805_filterd.fq.gzペアエンドの場合は、-i, -Iオプションで入力ファイル、-o, -Oオプションで出力ファイルを指定します。
fastp -i SRR24920875_1.fastq -I SRR24920875_2.fastq -o SRR24920875_1_filterd.fq.gz -O SRR24920875_2_filterd.fq.gz
fastpのオプション
fastpの詳細なオプションは公式のドキュメントを参照ください。いくつかのオプションの使い方はこの後説明します。fastp –helpコマンドで確認することも可能です。
出力データの確認
SRR24920875のペアエンドのFASTQファイルを実際にfastpで前処理してみましょう!
ターミナルで以下のコマンドを実行します。
docker container run --rm -v $PWD:/output -w /output \
bioinfosite/fastp:v0.23.4 fastp -i SRR24920875_1.fastq -I SRR24920875_2.fastq \
-o SRR24920875_1_filterd.fq.gz -O SRR24920875_2_filterd.fq.gz正常に実行されると、結果のレポートがfastp.htmlファイルで出力されます。このファイルを開くと、ブラウザ上で結果を確認できます。
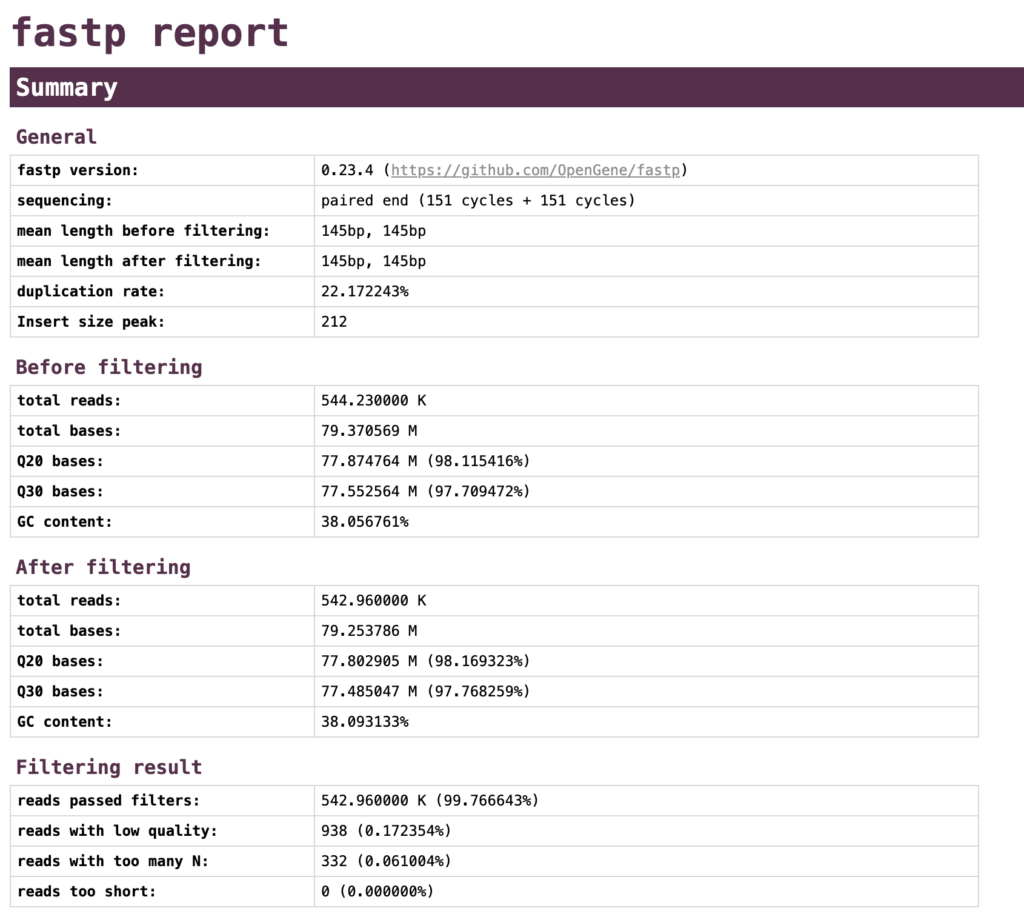
Summary
Summaryでは、フィルタリング前後での概要のレポートが記載されています。
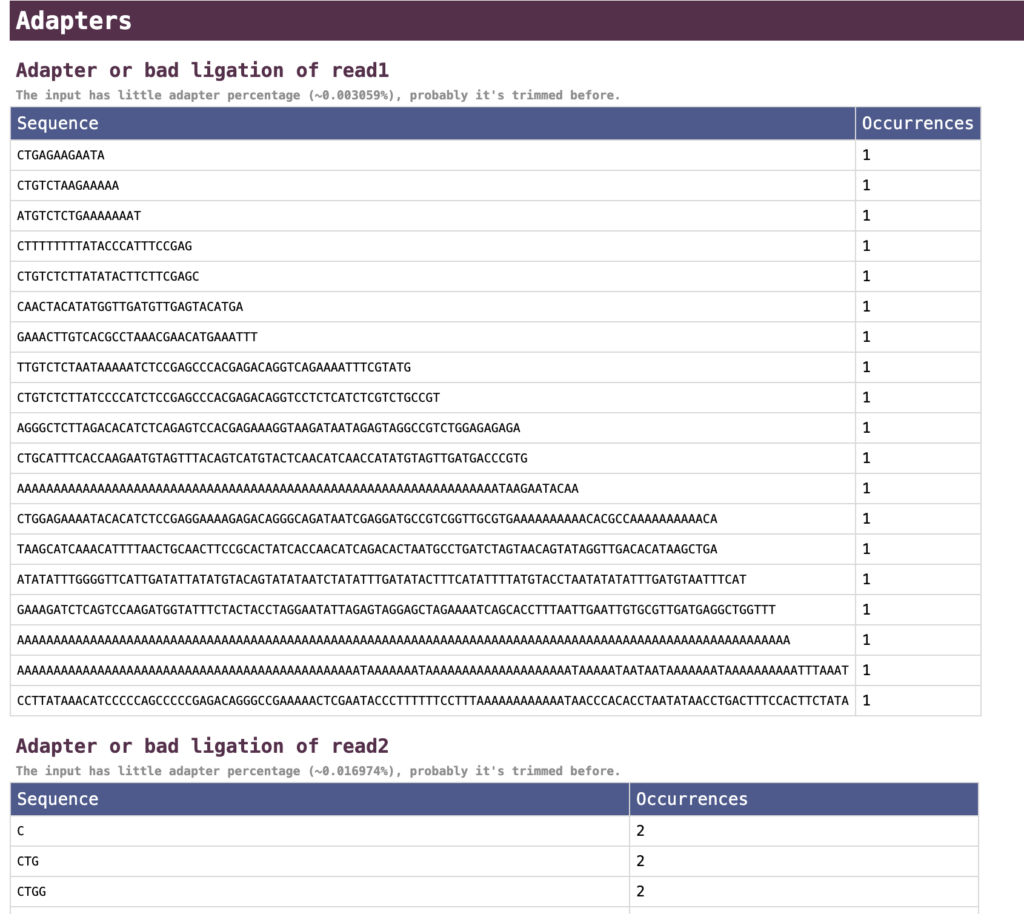
アダプター
アダプターに関する情報。fastpではアダプターを自動で検出し、トリミングしてくれます。今回の結果では、アダプターとして検出された配列の割合が少ないため、事前にトリミングされていることがfastpにより推測されています。
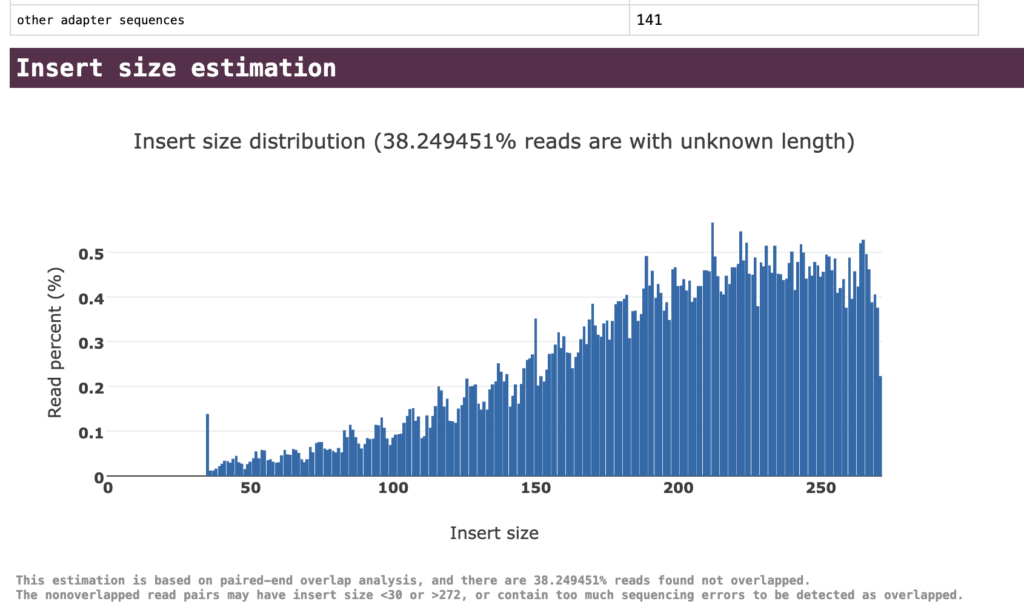
インサートサイズの分布。
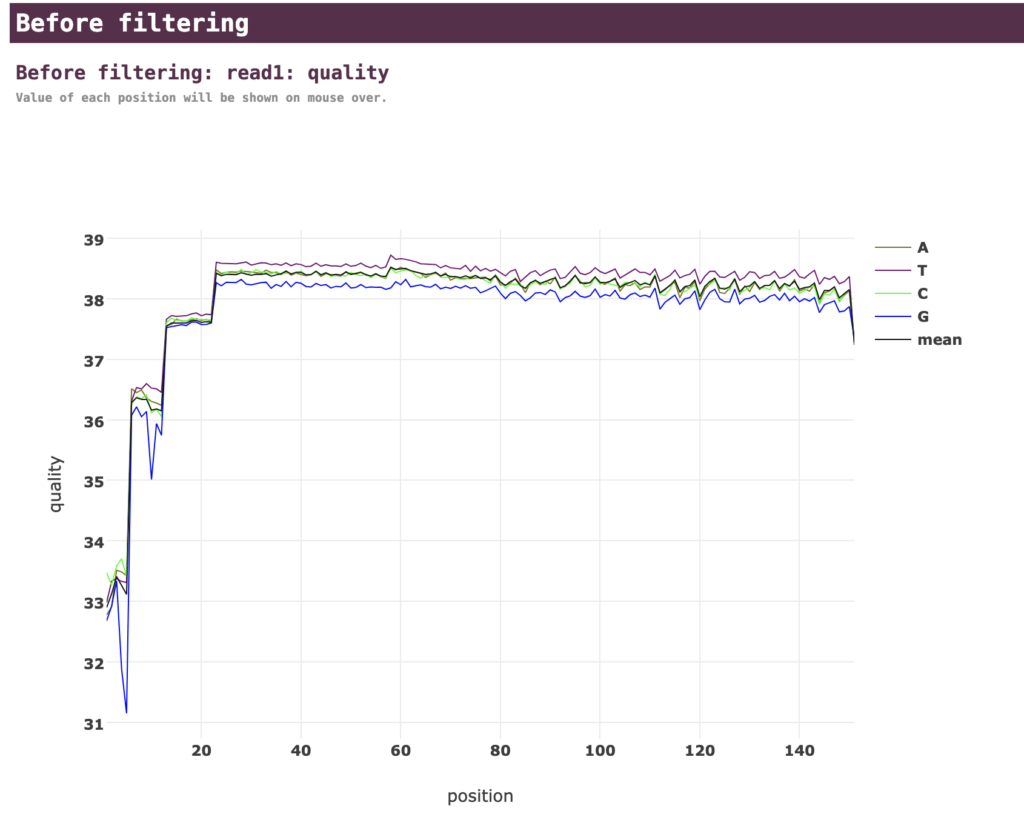
クオリティースコア
リードの各位置における塩基のクオリティースコアが示されます。このサンプルでは、すべての位置でクオリティースコアが30以上(読み取り精度99.99%以上)となっており、品質が高いことが読み取れます。後の解析に応じて、どれぐらいのクオリティースコアが必要かを決めます。ここでは示しませんが、後の方にフィルタリング後のクオリティースコアの分布の結果もあります。
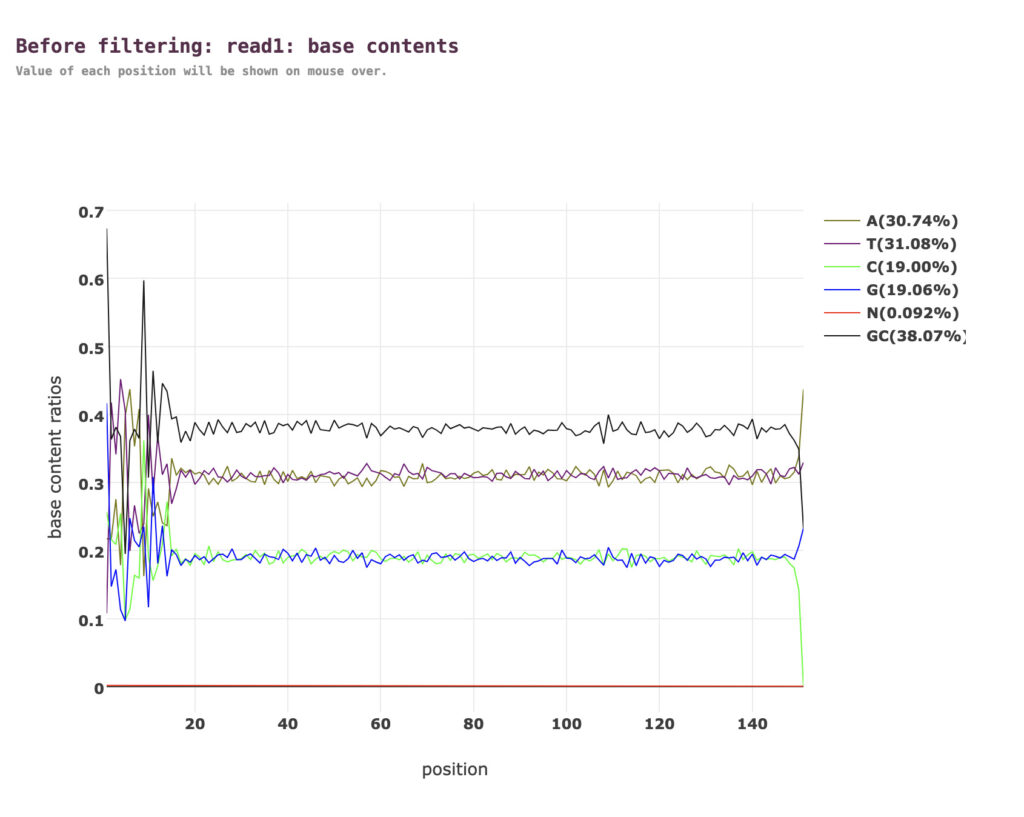
塩基割合
フィルタリング前のリードの各位置におけるATGCの割合が示されます。塩基の割合に偏りがないかを確認します。ここでは示しませんが、塩基割合も後の方にフィルタリング後の結果を表示されます。
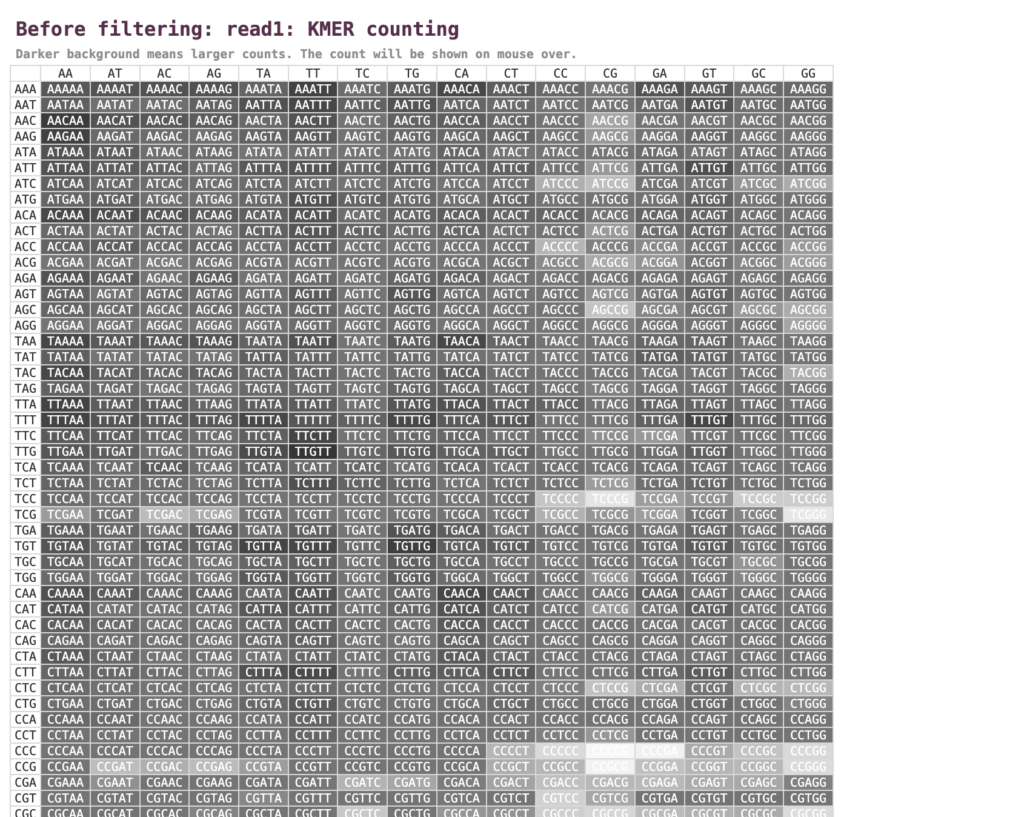
kmerカウント
頻度が高いkmerが、色が濃く示されています。
オプションを含めた使い方
SRR24464805のシングルエンドのデータをfastpのオプションを含めて実行してみます。
docker container run --rm -v $PWD:/output -w /output \
bioinfosite/fastp:v0.23.4 \
fastp -i SRR24464805.fastq -o SRR24464805_filterd.fq.gz \
-w 4 -f 1 -t 1 -l 30 -x -h report.html -j report.jsonオプションの説明は下記の通りです。
-f, –trim_front1 : リードの前方から何塩基トリミングするか。デフォルトは0
-t, –trim_tail1 : リードの後方から何塩基トリミングするか。デフォルトは0
-l, –length_required : この長さより短い塩基は除去する。デフォルトは15
-x, –trim_poly_x : 3’末端のポリXトリミングをおこなう
-h, –html : 結果htmlファイルの名前。デフォルトはfastp.html
-j, –json : 結果jsonファイルの名前。デフォルトはfastp.json
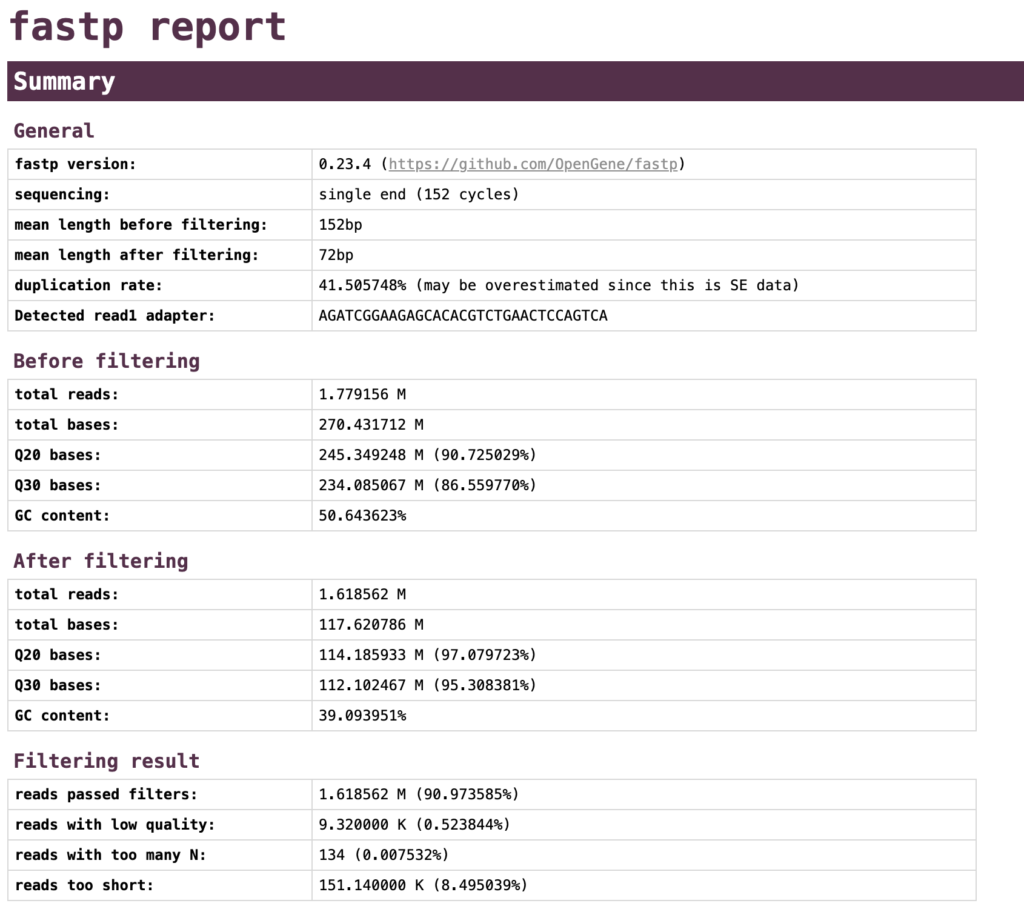
report.htmlをブラウザで開いて、結果を確認します。
おわりに
本記事では、FASTQファイルの前処理を一括でおこなうツールであるfastpの使い方を紹介しました。
結果もHTMLファイルのレポートとして出力されるので使いやすいです。結果をみながらオプションを変えて、フィルタリングの方法を変更するなど色々試してみてください。







