


本記事では、PyDESeq2を用いて、発現変動遺伝子を解析する方法について解説します。
PyDESeq2とは
PyDESeq2の前に、DESeq2について説明します。
DESeq2とは
DESeq2 は、次世代シーケンシング(NGS)データ、特に RNA シーケンシング(RNA-Seq)データの差次的発現解析を行うために設計されています。
主に遺伝子の発現量が条件や処理によってどのように変化するかを解析するために使われます。
https://bioconductor.org/packages/release/bioc/html/DESeq2.html
主な機能
- 正規化: DESeq2 は、サンプル間のライブラリサイズの違いを補正するために、データの正規化を行います。これにより、異なるサンプル間で発現量の比較が可能になります。
- 差次的発現解析: DESeq2 は、統計的な手法を用いて、異なる条件下での遺伝子の発現の変化を検出します。これには、各遺伝子の発現量の変化に対して
p-valueを計算し、統計的に有意な変化を検出します。 - 統計モデルの使用: DESeq2 は、カウントデータの変動をモデル化するために負の二項分布を使用します。これにより、発現量の変動を考慮に入れた厳密な差次的発現解析が可能になります。
- 多重検定の補正: DESeq2 は、多重検定の問題に対処するために、False Discovery Rate (FDR) の補正を行います。これにより、偽陽性のリスクを低減します。
以上のように、DEseq2は遺伝子発現量解析において不可欠な役割を果たしますが、使用するためにはR プログラミング言語が必要となります。
Rに抵抗がある方のために、この機能をpythonで実行する方法を解説します。
PyDESeq2とは
PyDESeq2 は、DESeq2のpythonバージョンであり、DESeq2の機能を pythonで実行できるようにしてくれています。
下記のリンクから、PyDESeq2の公式ドキュメントにアクセスできます。
PyDESeq2のインストール
pip install pydeseq2PyDESeq2の使い方
example_dataを使って手順を説明します。
データのインポート
PyDESeq2には、2種類の入力データが必要になります。
- 行がサンプル、列が遺伝子のカウントデータ。pandasのDataFrame形式。
- メタデータ。行がサンプルで、サンプルのラベルを記載する。pandasのDataFrame形式。
具体例として、以下のコマンドでpydeseq2の中に含まれるサンプルデータを読み込みます。
from pydeseq2.utils import load_example_data
counts_df = load_example_data(
modality="raw_counts",
dataset="synthetic",
debug=False,
)
metadata = load_example_data(
modality="metadata",
dataset="synthetic",
debug=False,
)counts_dfが①のデータ、metadataが②のデータに対応します。
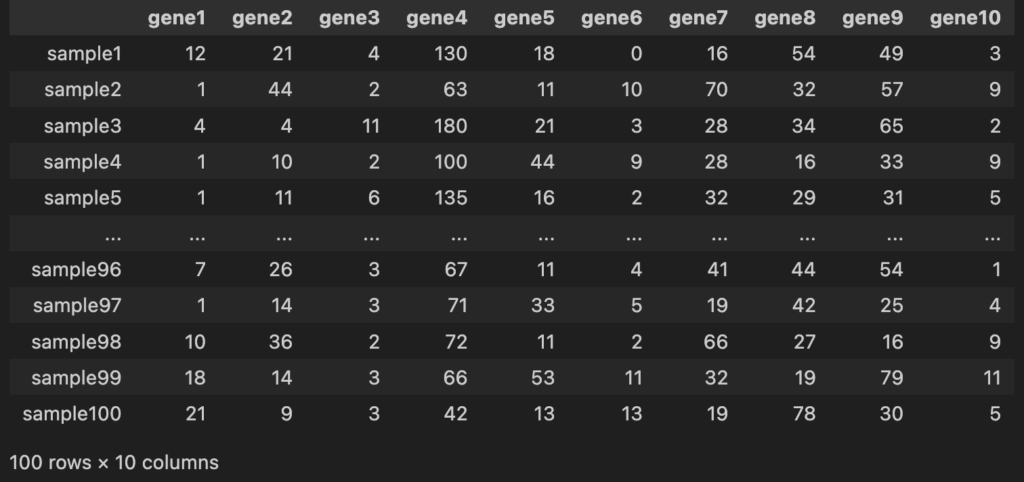
counts_dfは、100サンプル(行)x 10遺伝子(列)のデータフレームとなっており、カウント数がデータとして入っています。
counts_dfの中身
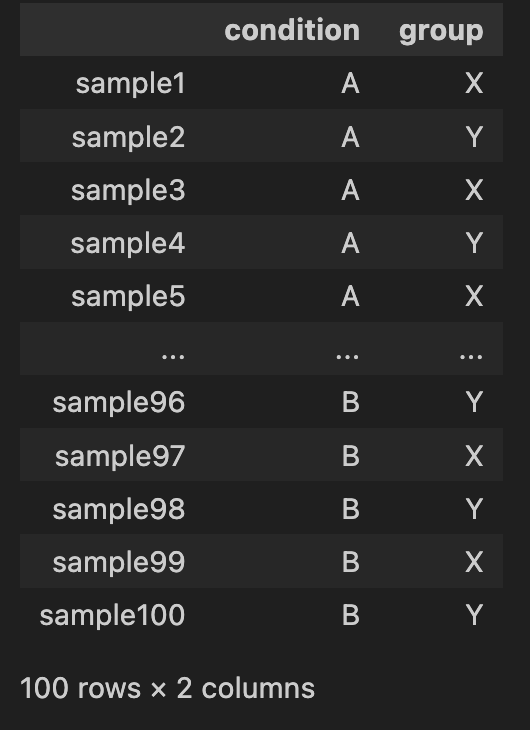
metadataは100サンプルのcondition(A or B)とgroup(X or Y)が入っているデータとなっています。今回の記事では、metadataのgroupは使わず、conditionのみを使います。
metadataの中身
自分で取得したデータを解析する場合には、counst_dfとmetadataのように2つのデータを上記の形式で準備します。
データのフィルタリング
全てのサンプル分を合計したカウント数が 10 未満の遺伝子を除外します。(このサンプルデータには、遺伝子が10個しかないので、そのような遺伝子はありませんが、実データの場合は除外される遺伝子があると思います。)
genes_to_keep = counts_df.columns[counts_df.sum(axis=0) >= 10]
counts_df = counts_df[genes_to_keep]
発現変動遺伝子解析
ここまで準備できたら、conditionのAとBの違いによる遺伝子発現量の差異を解析していきます。
まず、先ほど準備したカウントデータとメタデータからDeseqDataSetオブジェクトを作成します。
import os
import pickle as pkl
from pydeseq2.dds import DeseqDataSet
from pydeseq2.default_inference import DefaultInference
from pydeseq2.ds import DeseqStats
inference = DefaultInference(n_cpus=4)
dds = DeseqDataSet(
counts=counts_df,
metadata=metadata,
design_factors="condition",
refit_cooks=True,
inference=inference,
)作成したDeseqDataSetオブジェクトをもとに分散と log-fold change (LFC) を推定します。
dds.deseq2()出力結果
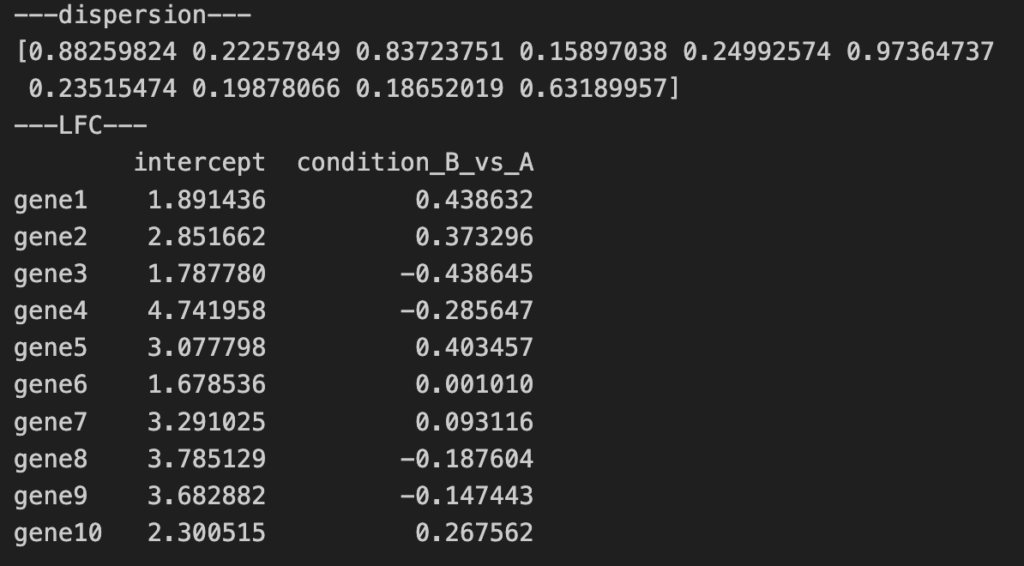
推定した分散とLFCを確認してみます。
# 分散
print("---dispersions---")
print(dds.varm["dispersions"])
# log-fold change
print("---LFC---")
print(dds.varm["LFC"])出力結果
分散と LFC が計算できたら、遺伝子発現量の変化が統計的に分析します。
PyDESeq2 は、Wald 検定を用いて p 値および調整済みp値を計算します。
stat_res = DeseqStats(dds, inference=inference)
stat_res.summary()
出力結果
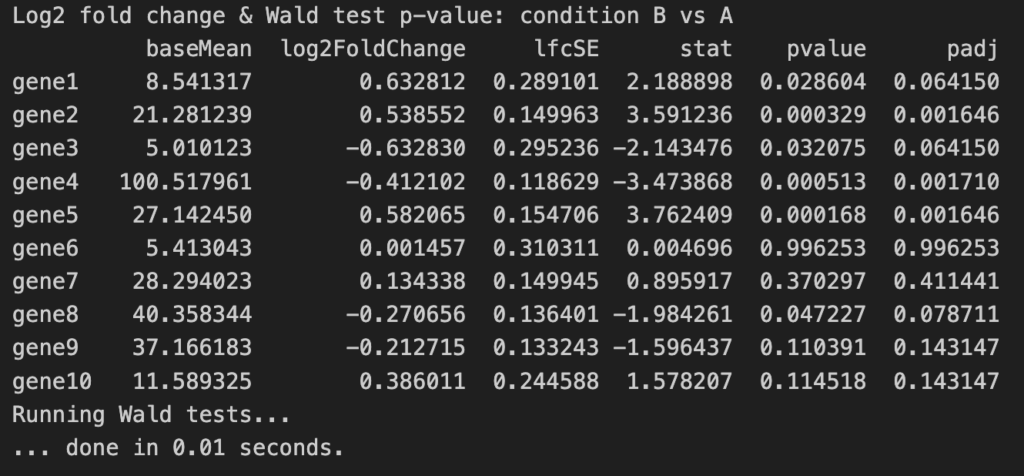
出力結果の見方を簡単に説明します。
- baseMean: 遺伝子の平均カウント値。全サンプルの遺伝子ごとの平均発現量を示しています。
- log2FoldChange: condition BがAに対して何倍変化したか、のlog2をとったもの。例えば、2倍増加した場合は1になる。
- lfcSE: log2FoldChangeの標準誤差。
- stat: Wald統計量の値。
- pvalue: Wald検定のp値。
- padj: 補正後のp値。多重検定の補正した値。
遺伝子発現が有意に変動しているかどうか、padjの値を確認し、発現変化量はlog2FoldChangeで確認できます。
以下のコードで、MAプロットを簡単に描画できます。
stat_res.plot_MA(s=20)出力結果
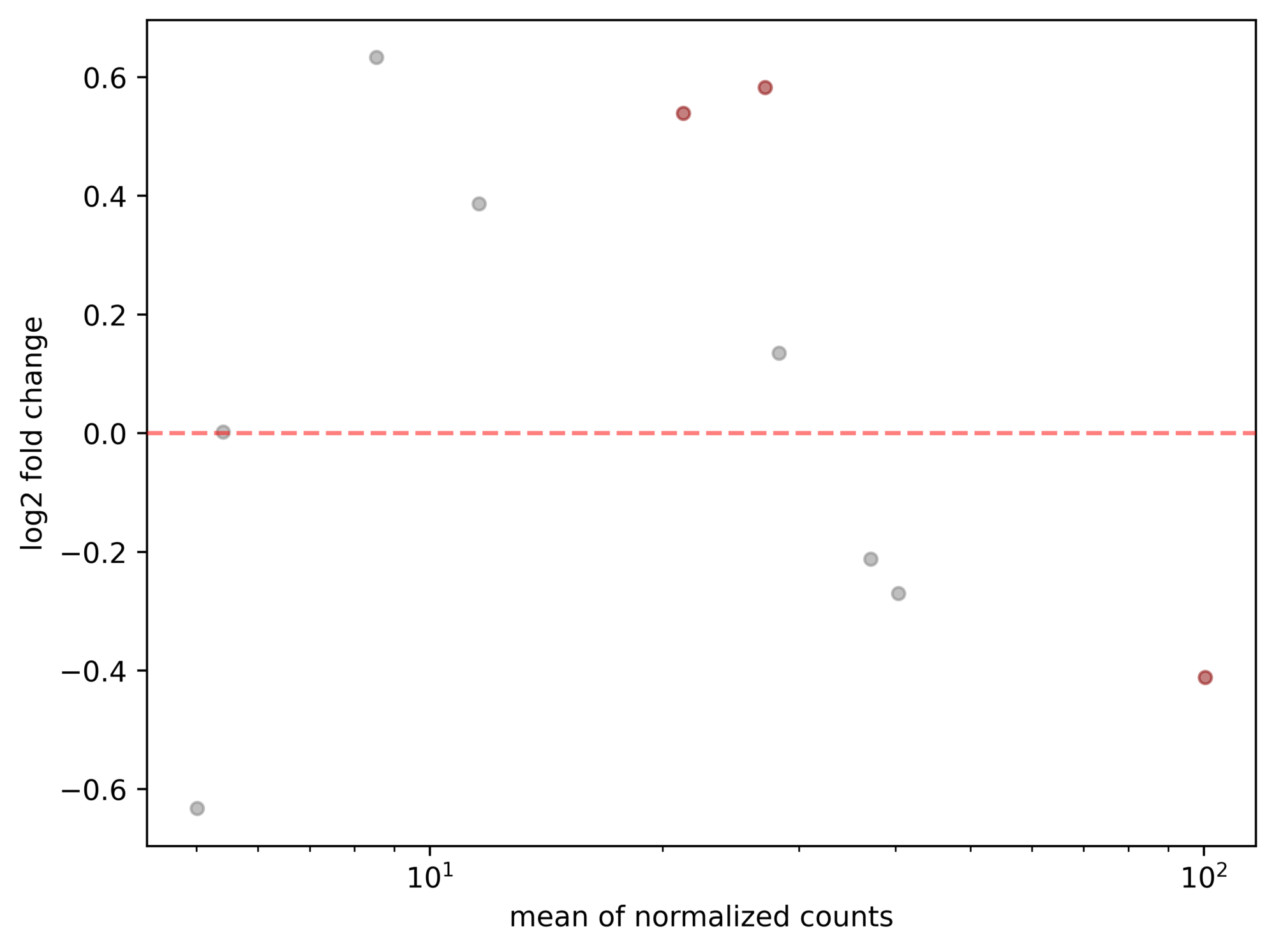
今回のサンプルデータセットでは、遺伝子数が10個と少ないため、さみしい感じのMA plotになっていますが、簡単にプロットすることができました。
おわりに
この記事では、PyDESeq2を用いて発現変動遺伝子を解析する方法を解説しました。
今回の記事では、サンプルデータセットを用いて解析の流れを解説しましたが、ぜひ、自分の興味のあるデータを解析してみてください。


